
Nga laboratori te pacienti – rruga drejt miratimit të një medikamenti në Bashkimin Europian (BE), Islandë, Norvegji dhe Lihtenshtain.
Rrugëtimi i një medikamenti, para se të arrijë te pacienti, nis me kërkimin shkencor rreth tij dhe përpjekjes për ta zhvilluar atë. Gjatë kësaj periudhe dhjetëra mijëra substanca testohen nga kompani farmaceutike apo bioteknologjike, si dhe nga personel mjekësor e akademik, por një numër i vogël rezulton i përshtatshëm për t’u testuar më pas në njerëz. Në vijimësi një numër edhe më i vogël sjell rezultate të mira në studimet e gjera klinike dhe në përfundim një numër akoma dhe më i vogel i substancave arrijnë të miratohen për përdorim, me qëllim përfitime shëndetësore te/nga pacientët.
Pasi kompanitë farmaceutike apo bioteknologjike zhvillojnë apo zbulojnë substancën e përshtatshme, ata kanë mundësi të aplikojnë për një këshillim shkencor tek autoritetet përkatëse, si për shembull tek Agjensia Evropiane e Barnave (European Medicines Agency, EMA). EMA e ofron këtë këshillim shkencor, me qëllim mbështetje për një zhvillim të shpejtë dhe me cilësi të lartë të medikamenteve efektive dhe të sigurta për pacientin. Procedura që një medikament ndjek nga nisja për zhvillimin e tij deri te përdorimi nga ana e pacientit është përshkruar më poshtë (Fig. 1).
Kjo procedurë mund të përmblidhet për thjeshtësi në dy faza kryesore, ajo e para miratimit të medikamentit dhe ajo e pas miratimit (post-miratimit) të tij.
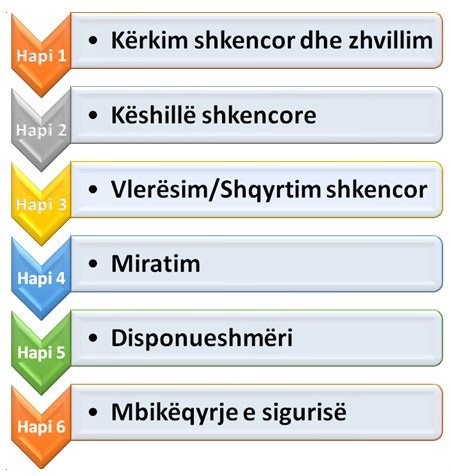
Fig. 1 Procedura që një medikament ndjek nga zhvillimi në laborator te pacienti
Faza e para miratimit të një medikamenti në BE:
EMA ofron udhëzime rregullatore për kompanitë të cilat kanë mbledhur mjaftueshëm rezultate nga kerkimet e tyre shkencore, në lidhje me medikamentin kandidat. Kjo kryhet disa muaj para aplikimit për miratim, duke u siguruar kompanive ndihmë për të përputhur lehtësisht dosjen e tyre aplikuese me kërkesat ligjore dhe rregullatore të Agjensisë. Kjo gjë sjell shmangien e vonesave të panevojshme.
Hapat para pranimit të aplikimit për miratim të një medikamenti në BE janë:
1. Kërkimi shkencor rreth substancës.
2. Studimet klinike të ndarë në 3 faza.
- Faza e parë e studimeve klinike përfshin mbledhjen e informacionit nga artikuj apo libra shkencorë, eksperimente dhe testime laboratorike. Në këtë fazë përcaktohet sasia e substancës që tolerohet nga pacientët si dhe doza toksike e saj.
- Faza e dytë e studimeve klinike përfshin testimin e medikamentit/ substancës në një numër të kufizuar pacientësh/individësh (disa qindra).
- Faza e tretë e studimeve klinike përfshin testimin e medikamentit/ substancës në një numër të gjerë (dhjetëra/mijëra) pacientësh/ individësh. Kjo është dhe faza përfundimtare klinike para se kompania të dorëzojë aplikimin për miratim të barit/substancës në BE, Islandë, Norvegji dhe Lihtenshtain.
3. Mbledhja e dokumentacionit të plotë dhe përgatitja e dosjes së aplikimit për një miratim të mundshëm (licensim). Dosja përmban gjithë informacionin me të dhënat e disponuesheme rreth medikamentit/ substancës (për shembull rezultate nga të gjitha studimet klinike dhe laboratorike, përmbledhje artikujsh, testime rreth menyrës së prodhimit dhe cilësisë, etj). Këto informacione dhe të dhëna duhet të paraqiten në mënyrë të strukturuar sipas nivelit të standarteve evropiane per miratim, të përcaktuara nga EMA.
Faza e miratimit të medikamenteve në BE:
Kompanitë depozitojnë dosjen, e cila përmban informacionin e nevojshëm rreth zhvillimit, testimit dhe cilësisë së medikamentit, e cila më pas pranohet dhe shqyrtohet nga EMA. Qëllimi i EMA-s është përcaktimi final nëse medikamenti është i sigurtë, efektiv, me cilësinë e duhur dhe i përshtatshëm për përdorim te pacientët në tregun Evropian. Shqyrtimi shkencor i aplikimit dhe rekomandimi për të lejuar tregëtimin e medikamentit ne BE bëhet nga komiteti shkencor i EMA-s për Produkte Mjekësore për Përdorim Njerëzor (CHMP). Ky komitet organizon mbledhje mujore, ku marrin pjesë perfaqësues nga të gjitha vendet e Bashkimit Evropian si dhe përfaqesues nga Islanda, Lihtenshtain dhe Norvegjia. Tre të fundit nuk kanë te drejtë votimi. Në mbledhjet mujore komiteti diskuton dhe merr vendime në lidhje me rekomandimin shkencor (opinionin) pozitiv ose negativ. Opinioni pozitiv nënkupton që ky komitet favorizon miratimin, ndërkohë që opinioni negativ nuk e favorizon. Rekomandimi vendoset duke arritur një marreveshje ndërmjet pjestarëve të komitetit të cilët arrijnë në konsensus, në të kundërt, vendimi merret nëpërmjet votimit, ku shumica e votave vendos rezultatin përfundimtar.
Faza e pas miratimit:
Komisioni Evropian jep vendimin ligjor për miratimin e një medikamenti duke u bazuar te rekomandimi pozitiv i lëshuar nga EMA. Pas këtij miratimi, medikamenti bëhet i disponueshëm për t’u hedhur në treg (shpërndahet në farmaci dhe/ose në qëndra spitalore). Njëkohësisht pas hedhjes së tij në treg, fillon faza e post-autorizimit e cila ka për qëllim mbikëqyrjen e sigurisë. Kjo fazë është shumë e rëndësishme për të garantuar sigurinë e medikamentit, pasi gjatë studimeve klinike nuk është e mundur të zbulohen të gjitha efektet anësore e në veçanti ato afatgjata. Prandaj mbikëqyrja gjatë fazës së post-autorizimit dhe gjatë gjithë ciklit (lifecycle) të medikamentit është po aq i rëndësishëm sa faza e para miratimit. Përfitimet dhe rreziqet e një medikamenti shqyrtohen herë pas here dhe periodikisht për të parë nëse raporti përfitim/rrezik mbetet gjithmonë në favor të përfitimeve. Në qoftë se kjo balancë ndryshon në favor të rrezikut atëherë medikamenti detyrohet të tërhiqet nga tregu (farmacitë/qendrat spitalore) evropian dhe miratimi i tij anullohet në të gjitha vendet e BE, Islandë, Norvegji dhe Lihtenshtain.
Referencat:
4. https://www.ema.europa.eu/en/from-lab-to-patient-timeline
6. https://eur-lex.europa.eu/legal-content/EN/TXT/HTML/?uri=CELEX:02001L0083-20121116&from=EN
8. https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=CONSLEG:2004R0726:20130605:EN:PDF
Lexo edhe: Si po aprovohen vaksinat ANTI-COVID-19 në BE



